Hauptinhalt
Topinformationen

Conformational dynamics of disease-related proteins and protein complexes (Klare)
"... everything that living things do can be understood in terms of the jigglings and wigglings of atoms." (Feynman, Leighton et al. 1963)
Proteins are highly dynamic entities. They exhibit conformational fluctuations where side chains adopt different orientations and secondary structure elements as well as whole domains can move against each other. Motion is required to bind or release ligands, substrates, nucleic acids or other proteins, or to transduce signals. We apply different biophysical techniques, foremost site-directed spin labelling (SDSL) in combination with electron paramagnetic resonance (EPR) spectroscopy, and molecular dynamics (MD) simulations to characterize the conformational ensemble that represents a protein’s native state, and to unravel how protein structure and dynamics relate to biological function and dysfunction. Current subjects of our investigations are several so-called G proteins activated by dimerization (MnmE, hGBP1, Roco proteins (incl. the human “Parkinson kinase” LRRK2)), small G proteins (Ras), different channels (e.g. VDACs) and transporters, and the archaeal SRII/HtrII photoreceptor/transducer signaling complex.
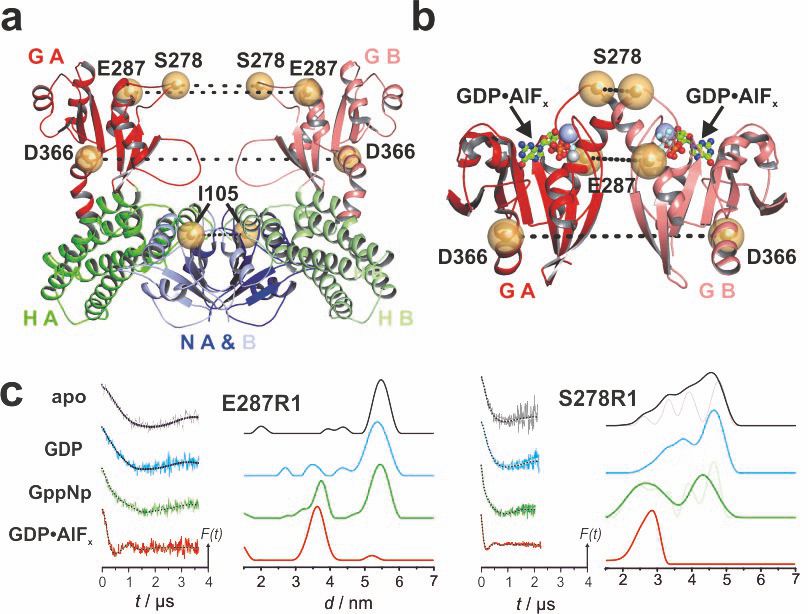
Figure: Spin label sites in the tRNA-modifying MnmE dimer. Position of residues that were mutated to Cys and spin labeled (yellow spheres), with dashed lines indicating distances between residues in the open (A) and closed (B) conformation. (C) DEER characterization of nucleotide-dependent domain movements of MTSSL labelled MnmE (E287R1, D366R1, S278R1, and I105R1). Left panels: background corrected dipolar evolution data. Right panels: distance distributions obtained by Tikhonov regularization. Excerpt from Meyer et al. (2009), doi.org/10.1371/journal.pbio.1000212.
Molecular Mechanisms of Biological Energy Conversion and Their Evolution (Mulkidjanian)
We try to understand the molecular mechanisms of energy conversion by enzymes. Initially we studied these mechanisms by using spectrophotometric and electrometric techniques that enable to trace subnanoscopic displacements of electric charges inside membrane proteins and at their surface with a resolution of up to 0.1 nm in space and up to 100 ns in time. More recently we became interested in the evolution of these mechanisms. We combine the tools of bioinformatics and comparative genomics with kinetic modeling, electrostatic calculations and molecular dynamics simulations. Such an evolutionary biophysics approach allowed us not only to reconstruct the origin and evolution of key membrane energy-converting enzymes (ATP synthase, cytochrome bc1 complex, NADH:ubiquinone oxidoreductase) and of the membrane bioenergetics as a whole, but also to identify several new enzyme families and biochemical pathways, see the publication list.
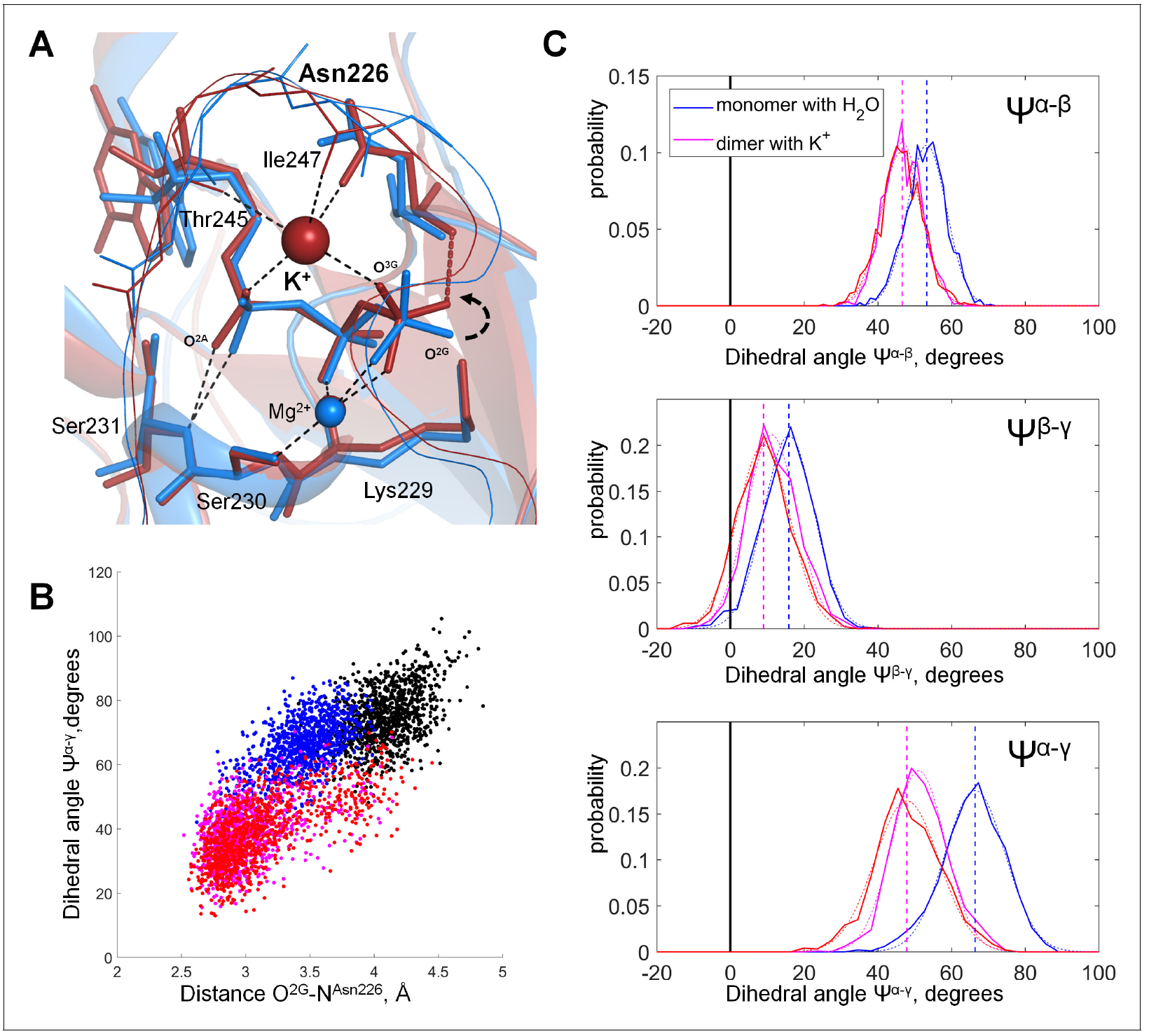
Figure from: D. N. Shalaeva, D. A. Cherepanov, M. Y. Galperin, A. V. Golovin, A. Y. Mulkidjanian (2018) Evolution of cation binding in the active sites of P-loop nucleoside triphosphatases in relation to the basic catalytic mechanism, eLife 2018;7:e37373, DOI: doi.org/10.7554/eLife.37373.


